Focus on: Cryo-electron microscopy
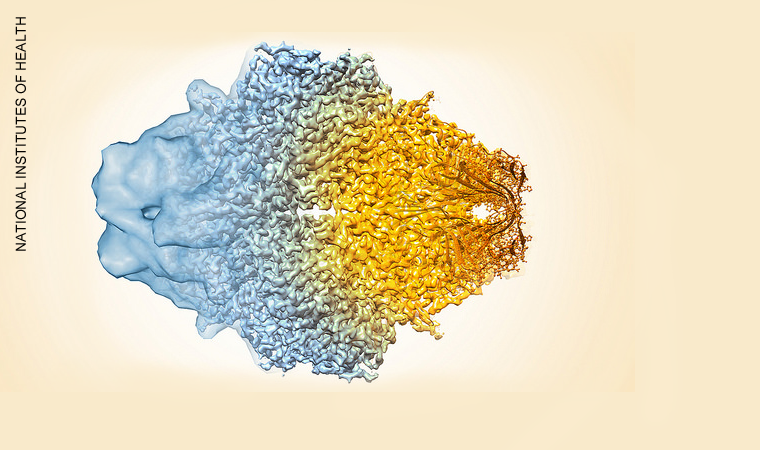
The cool technique revealing molecular structures in thrilling detail
The Biologist 63(5) p32-33
Cryo-electron microscopy, or cryo-EM, uses electrons to study samples frozen at cryogenic temperatures. In the past five years, it has become the go-to technique for studying the structural arrangement of biological samples, sometimes achieving near atomic resolution.
According to an article in Nature last year, cryo-electron microscopes are “sending tremors through the field of structural biology…in the past three years, they have revealed exquisite details of protein-making ribosomes, quivering membrane proteins and other key cell molecules, discoveries that leading journals are publishing at a rapid clip”.

Molecular movies
Large macromolecules and subcellular structures have proven particularly good targets for imaging with cryo-EM. Some of the most exciting advances in the field have been in determining the 3D structure of ribosomes, proteins and viruses, almost to the atomic scale. Stitching hundreds of thousands of images together can even help reveal how structures within the molecules move, such as the rotating elements of motor-proteins and the complex working parts of the enzymes that oversee the transcription of genes.

How it works
Single-particle cryo-electron microscopy
Freezing
The sample is applied to a grid, which is then frozen in liquid ethane and stored in liquid nitrogen. Freezing must be fast enough to prevent the water present forming ice crystals. Ice lattices will not only absorb the electron beam and obscure the image, but are likely to damage the sample's structures. If the sample is frozen quickly enough, water solidifies as an amorphous solid (vitreous ice) and does not crystallise.
Recording
Images of multiple copies of the molecule (or other material, such as viruses) suspended in random orientations in vitrified ice are recorded, based on the interactions of the matter with a beam of electrons. Newly developed 'direct electron detectors' record better-quality images than older digital camera-style imaging.
Data processing
The structure of the sample is determined by combining the different views of the molecule into a 3D model. The final image, or density map, is sometimes the average of tens or hundreds of thousands of particle images. Hundreds of thousands of frames per second are recorded to help trace the movement of particles and so reduce loss of resolution.
PROS
A major advantage of cryo-EM over x-ray crystallography is that the molecule of interest does not have to be crystallised. Some proteins or important macromolecules simply can not be crystallised; others have their structures irreversibly changed by crystallisation.
Proteins can be seen in all conformations they adopt, unlike in x-ray crystallography, in which only a single structure can be determined.
Unlike conventional electron microscopy, cryo-EM samples are not dehydrated or stained, which means their structure remains close to the true shape of the hydrated structure in their native environment, and there are no false shapes created by the stain.
Freezing samples minimises the radiation damage that samples can suffer as a result of electron irradiation. Frozen samples are also less likely to be damaged by the low pressure/vacuum conditions of the electron microscope.
CONS
Due to a lack of stain and therefore a lack of contrast, images often have a very low signal-to-noise ratio, requiring highly advanced detection hardware and image processing.
Specimen preparation can be tricky – not only to optimise ice thickness, but also to optimise particle distribution. Sometimes proteins adopt preferred orientations that make 3D reconstruction impossible.
The most advanced cryo-EM equipment remains extremely expensive. The development of centralised facilities may be able to help increase access to cryo-EM equipment.
